
機能詳細1 分子モデリング・シミュレーション
MOEの分子モデリング環境には、分子を表示・操作・管理する機能と、多彩な分子構築ツールが搭載されています。構築した分子系に対して、ポテンシャルエネルギー計算や安定構造の予測、熱運動の解析など分子設計のための基本となる精度の高い分子シミュレーションが可能です。
分子モデリング環境
分子構築
低分子、ペプチド、糖鎖、DNA、RNA、合成高分子を構築するツールを備え、各種分子が簡単に構築できます。非天然のアミノ酸や核酸の構築にも対応しています。2Dスケッチャーと連携させた対話的な構造構築や、コピー&ペーストによる構築、分子名やCAS登録番号からの構築にも対応しています。周期境界条件の設定や溶媒分子/カウンターイオンを配置する機能を持ち、バルクモデルの構築も行えます。
分子構造の表示と出力
生体高分子、低分子およびそれらの複合体の3次元表示が可能です。リガンドの2次元表示を同一ウィンドウ内に表示できます。ラベルとして、分子/残基/原子/実験データなど様々な情報を自由に付与できます。プレゼンテーションや論文用に、焦点の合う範囲を狭めたぼかし効果や模式図的な表現などの描画効果を適用した画像、動画が出力できます。赤ーシアンなどのアナグリフ眼鏡を始めとした様々な立体視に対応しています。パワーポイントやウェブページに埋め込み可能な3Dグラフィックスや、3Dプリンター用の出力に対応しています。
相互作用表示
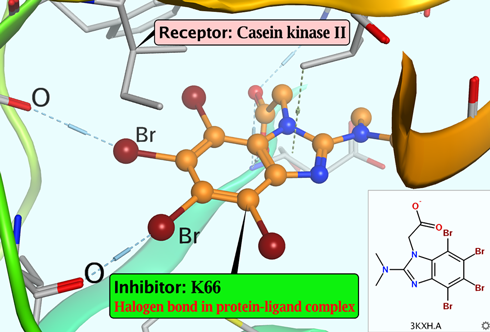
リガンドとタンパク質や核酸との複合体において水素結合、ハロゲン結合、CH-π相互作用、ファンテルワールス接触などの情報を表示できます。拡張Hückel法に基づいた電子状態計算により、リアルタイムに相互作用エネルギーの定量的な評価が行えます。
アミノ酸・塩基配列表示
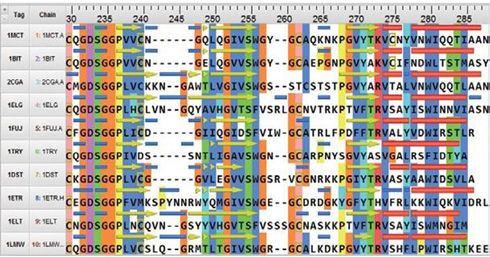
配列表示ウィンドウでは配列と立体構造を連携して操作することができます。タンパク質および核酸の配列アラインメントと重ね合わせに対応し、アラインメント後の一致度やRMSD値などで色分けできます。タンパク質においては、二次構造やジスルフィド結合のアノテーション、残基ごとの溶媒露出度などのスコアの表示ができます。
分子構造データベース
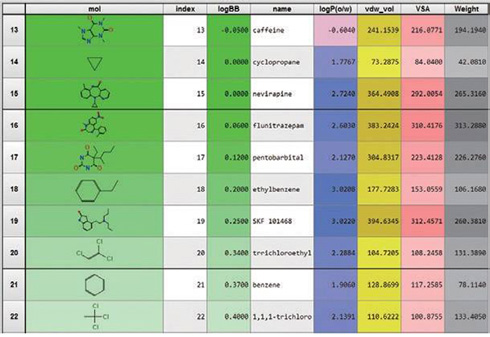
大量のデータを効率よく処理する独自のデータベース機能を搭載しています。タンパク質、低分子などの分子構造データ、数値データ、テキストデータを統一的に管理でき、約100万件の低分子を1GB 程度のファイルに保存できます。
分子シミュレーション
分子構造の前処理
- タンバク質立体構造の確認と修正
- タンパク質立体構造データに含まれる問題点を検出し、自動的に修正します。欠損している原子座標の補完やキャッピング、ジスルフィド結合の再構築、残基名の修正、部分電荷計算、水素原子付加状態の最適化、原子接触/シス型ペプチド結合の検出などが行えます。
- 水素原子付加状態の最適化(Protonate 3D)
- 分子系全体として最も安定な水素付加状態を探索し、温度、pH、塩濃度を考慮して標的タンパク質のイオン化状態、水素原子の位置、側鎖のフリップを最適化します。
- Protomers
- 指定のpHにおける低分子のイオン化状態や互変異性体の存在比率を計算します。
分子力学計算
分子力場に基づく、分子のポテンシャルエネルギー計算、リガンド受容体間の相互作用エネルギ一計算、構造最適化計算が行えます。MOE独自の力場として、Amber10:EHT、さらに核酸のパラメーターが改良されたAmber14:EHTが搭載されています。拡張Hückel法により自動的にパラメーターを作成することで、どのような原子タイプにも、適切なパラメーターが割りあてられます。その他、MMFF94s、Amber99、CHARM27、PFROSSTなど、様々な力場が搭載されています。溶媒効果として、距離依存誘電率や反応場モデル、一般化ボルンモデル(GB/VI) が利用できます。原子の固定、原子間拘束、配座固定が設定できます。

分子動力学計算と結合自由エネルギー計算
分子を熱運動させて、位置や速度、エネルギーなどについて動的経過を解析します。計算エンジンとしてNAMD※1、AMBER※1が利用でき、マルチコア、クラスター、GPUを利用した計算設定にも対応しています。AMBERの熱力学的積分法のインターフェースにより、相対的なリガンドー受容体間の結合自由エネルギーを簡単に計算できます。受容体と複数のリガンドの構造群から、計算に必要なインプット/スクリプトファイルを自動的に作成します。
配座解析
分子の最安定配座または複数の安定配座を予測する機能です。以下の手法を備えています。配座解析後に量子化学計算を利用した構造最適化計算を自動で行うこともできます。
| Systematic Search | 結合を順次回転させる網羅的な配座発生法。 |
|---|---|
| Stochastic Search | 結合をランダムに回転させる配座発生法。 |
| LowModeMD | 低周波振動モードに重み付けした分子動力学計算による配座発生法。低分子だけではなくペプチドやタンパク質のループ構造にも適用可能。 |
| Conformation Import | フラグメント単位での配座発生法。大量の化合物を高速処理。 |
| Torsion Profile | 低分子のねじれ角を段階的に変化させてエネルギーやMogul※1による統計値を出力。 |
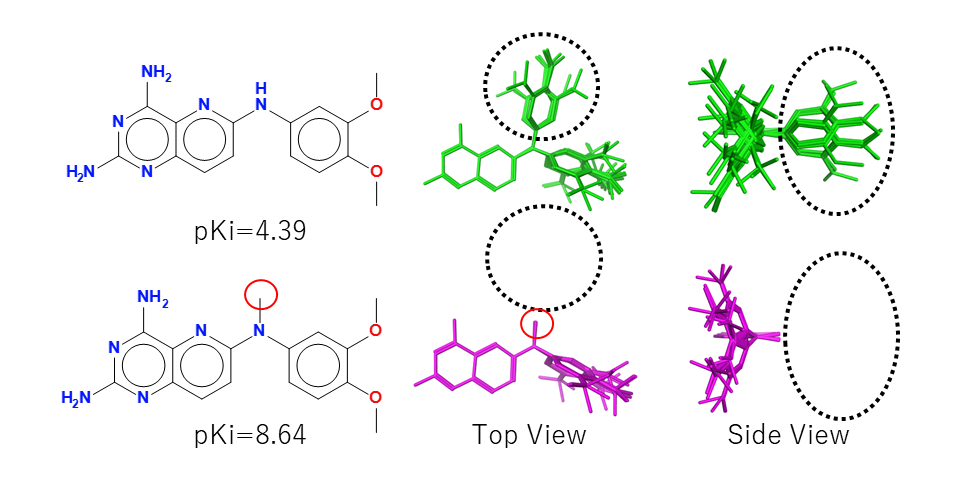
2種のDHFR阻害剤の配座解析の結果。下の分子はメチル基(赤丸)の存在により配座の自由度が制限(黒の破線)。
分子アラインメント
複数の低分子重ね合わせの機能が搭載されています。
| Flexible Alignment | 分子の配座を変化させながら分子特性を基準に複数の分子を重ね合わせ、重なりの度合いとひずみエネルギーで評価します。 |
|---|---|
| Molecule Superpose | テンプレートの分子に対して、複数の手法(分子構造全体、任意のフラグメント、SMARTS 、類似3原子、分子配座)に基づいてデータベースの分子を重ね合わせます。 |
量子化学計算インターフェース
MOEから外部の量子化学計算プログラム(MOPAC7、MOPAC2016※1、Gaussian※1、ADF※1、GAMESS※1) を利用して、分子軌道や電子密度、電子配置図の描画、生成熱の予測、部分電荷、構造最適化計算を行うことができます。GaussianではHessian行列を用いた多段階の構造最適化プロトコルが利用できます 。
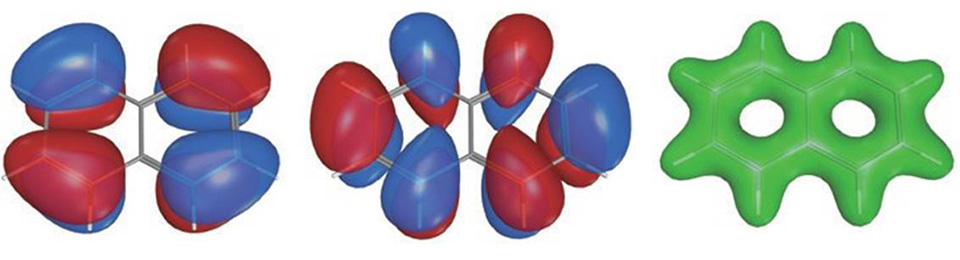
ナフタレンのHOMO(左)、LUMO(中)、電子密度(右)。
スペクトル解析
分子化合物の構造をMOEの配座解析機能により発生させ、量子化学計算ソフトウェアのGaussianと連携させることでNMRスペクトルやVCDスペクトルを予測します。計算結果と実験結果との重ね合わせや差分の表示が行えます。NMR実験により得られたケミカルシフトやJーカップリング、NOEデータを用いることで溶液中の分子配座の存在比を予測できます。
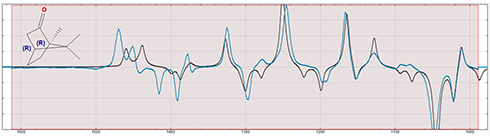
- ※1別途入手が必要
統合計算化学システムMOE に関するお問い合わせ
-
Webでのお問い合わせ
入力フォーム当社はセキュリティ保護の観点からSSL技術を使用しております。
-
お電話でのお問い合わせ
富士通コンタクトライン(総合窓口)
0120-933-200(通話無料)受付時間:9時~12時および13時~17時30分(土曜・日曜・祝日・当社指定の休業日を除く)